Vi tilrår at du alltid nyttar siste versjon av nettlesaren din.
Skal du forske? Dette må du vite
Retningslinjer for forsking i Helse Møre og Romsdal HF
I desse retningslinjene får du ein gjennomgang av dei formelle krava til forskingsprosjekt, frå planlegging til avslutning. Du bør setje deg inn i retningslinjene tidleg i prosessen. Retningslinjene bygg på dokumentet
«Regionale retningslinjer for (medisinsk og helsefagleg) forsking».
Presisering: I dokumentet blir omgrepet forskingsansvarleg brukt ved fleire høve. Forskingsansvarleg i Helse Møre og Romsdal HF er administrerande direktør, men oppgåvene knytt til dette ansvaret er delegert til fagdirektør og vidare til forskingssjefen. Til å hjelpe seg i rolla som forskingsansvarleg har forskingssjefen oppretta DAC-gruppa (sjå nærare omtale under avsnittet om intern forankring).
Retningslinjene inneheld ord og omgrep som kan vere nye og framande for mange. Derfor tilrår vi sterkt at du startar med å gjennom definisjonane i følgjande
ordliste.
Planlegging - korleis starte opp?
Eit nyttig verktøy i planleggingsfasen er «Helseforskingsportalen» som særleg gir hjelp til korrekt datahandtering.
Som prosjektleiar er du ansvarleg for planlegginga og den daglege drifta av forskingsprosjektet. Prosjektleiar gjennomfører prosjekt etter fullmakt frå forskingsansvarleg med eventuelle føringar eller vilkår frå denne. Prosjektleiar må i tillegg halde seg til relevante interne prosedyrar, vilkår frå eksterne godkjenningsinstansar som Regionale komitear for medisinsk og helsefaglig forskingsetikk, (REK), Statens legemiddelverk (SLV) og sentrale lover som
Personopplysingsloven, og
Helseforskingsloven. Prosjektleiar har også eit ansvar for at prosjektet blir gjennomført i tråd med behandlingsgrunnlaget (sjå eige avsnitt), oppgitt formål, og at alle prosjektmedarbeidarar overheld gitte vilkår, regelverk, og prosedyrar.
Kvalitetssikring og evaluering av pasientbehandlinga er ein del av helsetenestene. Helse- og omsorgsdepartementet sin rettleiar til helseforskingslova definerer kvalitetssikring som prosjekt, undersøkingar og evalueringar som har som føremål å kontrollere at diagnostikk og behandling faktisk gir dei tilsikta resultata. Generelt er forskjellen mellom forsking og kvalitetssikring slik (REK): Forsking handlar om å få ny kunnskap, om å finne ut kva som er eller vil bli beste praksis (kunnskapsbasert standardbehandling) – forskingsspørsmålet vil til dømes vere "kva er den mest effektive måten å behandle trykksår på?". Kvalitetssikring handlar om å finne ut om best praksis er følgd – spørsmålet vil til dømes vere "Korleis behandlar vi trykksår, og korleis kan dette samanliknast med akseptert beste praksis?". Forskjellen er ikkje absolutt. Ei nyttig tilnærming er å konsentrere seg om tre nøkkelspørsmål: 1. Er føremålet med prosjektet å forbetre kvaliteten på pasientbehandlinga lokalt? 2. Vil prosjektet innebere evaluering av praksis opp mot etablert standard? 3. Skal pasientane gjennomgå noko som elles ikkje er ein del av den normale rutinebehandlinga? Dersom svaret på dei to første spørsmåla er "ja" og svaret på det tredje er "nei", er prosjektet truleg kvalitetssikring, elles er det truleg forsking og skal normalt ha førehandsgodkjenning av REK.
Ved søknad til REK om dispensasjon frå teieplikt (unntak frå å be om pasientsamtykke) eller om vurdering av om prosjektet er meldepliktig til REK, må du vere merksam på følgjande: Dersom REK tydeleg definerer prosjektet som kvalitetssikring og ikkje gir dispensasjon frå teieplikt, står du att med eitt behandlingsgrunnlag for personopplysningar: § 26 i helsepersonellova. Ved bruk av dette behandlingsgrunnlaget, kan ein som hovudregel ikkje publisere vitskaplege artiklar frå prosjektet. Derfor bør du vurdere om du kan definere prosjektet ditt som «helsetenesteforsking» i søknaden og ikkje «kvalitetssikring». Er du usikker på om prosjektet er søknadspliktig til REK – ta kontakt med REK for rettleiing.
Sjå elles liste over prosjekt som normalt ikkje treng godkjenning frå REK.
Alle forskingsprosjekt i HMR skal ha godkjenning frå Data Access Committee (DAC-gruppa) før dei kan starte. Les meir om DAC-gruppa her.
Søknadsskjema til DAC (kun for tilsette) : Søknad er sendt ved ferdigstilling og lagring av søknadsskjemaet. DAC overtar då den vidare saksgangen.
Som prosjektleiar må du først sørge for at prosjektet er forankra internt før det kan starte. Det betyr å sikre støtte frå næraste leiar og at prosjektet er forankra hos ansvarleg klinikksjef. Dette gjeld for alle forskingsprosjekt, også helseforsking, som skal godkjennast av REK, anna forsking, kvalitetssikring og oppbygging av kvalitetsregister og av forskingsregister som tidlegare trengde konsesjon frå Datatilsynet. Forankringa skal vere dokumentert i søknadsskjemaet.
Dersom forskingsprosjektet ditt skal nytte seg av opplysingar frå pasientar, tilsette eller andre, vil prosjektet behandle personopplysingar. Før ein gjennomfører ei behandling av personopplysingar, må prosjektleiar dokumentere at han/ho har lov til å nytte personopplysingar til sitt føremål. Dette inneber å vise til eit såkalla behandlingsgrunnlag i personvernforordninga.
Behandlingsgrunnlaget må godkjennast av forskingsansvarleg og arkiverast i helseføretaket sitt saksbehandlingssystem. I HMR er det forskingssjefen som har fått delegert denne myndigheita, og forskingsprosjektet kan få behandlingsgrunnlag ved at søknad om godkjenning av forskingsprosjektet blir behandla og godkjend av DAC.
Du kan få hjelp til å utforme eit behandlingsgrunnlag av regional eller lokal personvernrådgivar (PVO) for forsking, eigen klinikk (dersom kompetanse) eller forskingstøtte i helseføretaket,
forsking@helse-mr.no
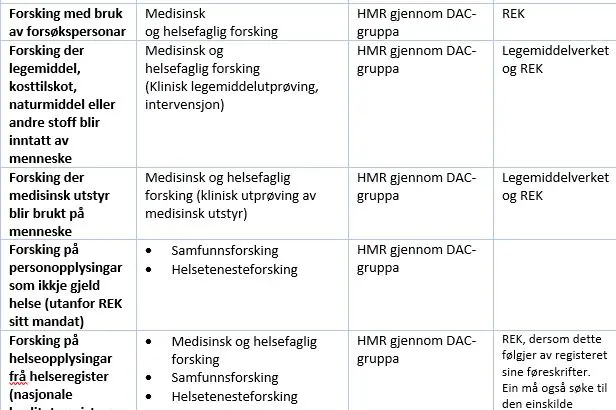
Oversikt over ulike typar forskingsprosjekt og kven som godkjenner behandlingsgrunnlag
[1] REK (Regionale komiteer for medisinsk og helsefaglig forskingsetikk). REK vurderer om forskingen blir drevet forsvarlig og gir etisk forhåndsgodkjenning av medisinsk og helsefaglig forsking
Nærare forklaring: sjå
lenke 1. Samtykke
2. Nødvendig for å oppfylle ein avtale
3. Nødvendig for å oppfylle ei rettsleg plikt
4. Nødvendig for å verne vitale interesser
5. Nødvendig for å utføre ei oppgåve av ålmenn interesse eller utøve offentleg mynde
6. Nødvendig for å vareta legitime interesser - interessevekting
Ei vurdering av personvernkonsekvensar (DPIA) avklarar om prosjektet etterlever relevant regelverk, og sikrar at personvernet til dei registrerte er ivaretatt. DPIA er særleg aktuelt i forskingsprosjekt som ikkje baserer seg på et informert samtykke. Ein DPIA skal dokumenterast etter personvernforordninga sine krav til innhaldet til ein DPIA. Der konklusjonen er at DPIA ikkje er nødvendig, skal også denne vurderinga dokumenterast. Les
Datatilsynets DPIA-rettleiar, kontakt personvernombod/ -rådgivar, eller sjå Datatilsynet sine nettsider for meir informasjon.
DAC vil gjennom saksbehandling vurdere om det bør utførast ein DPIA for det aktuelle forskingsprosjektet. Når ein finn det naudsynt å utføre DPIA, vil FIUK-seksjonen/DAC initiere utføringa og hjelpe prosjektleiar i gang.
På søknadsskjema under punkt 5.0 (fungerer kun på pc fra helseføretaka) registrerer ein informasjon som vil ligge til grunn for vurdering av DPIA. Dersom det alt er gjort DPIA på prosjektet (multisenterstudie), kan denne leggast ved og vere grunnlag for vurdering hos personvernombod/-rådgivar (PVO/PVR) og DAC.
REK Alle medisinske og helsefaglege forskingsprosjekt skal godkjennast av Regionale komitear for medisinsk og helsefagleg forskingsetikk (REK). Medisinsk og helsefagleg forsking er aktivitet utført med vitskapeleg metodikk for å skaffe ny kunnskap om helse og sjukdom. For medisinsk og helsefagleg forsking på menneske, humant biologisk materiale eller helseopplysingar, er det krav om etisk godkjenning av prosjektet frå REK før det kan starte. Meir informasjon om søknadsskjema, søknadsfristar og malar for samtykkeskjema finn du på REK sine sider.
REK skal førehandsgodkjenne: • Medisinske og helsefaglege forskingsprosjekt • Generelle forskingsbiobankar • Dispensasjon frå teieplikt i andre typar forskingsprosjekt
Sjølv om du har fått førehandsgodkjenning frå REK, er det viktig å hugse at dette åleine ikkje er nok som behandlingsgrunnlag. Ved behandling av personopplysingar i eit prosjekt er det alltid krav om eit behandlingsgrunnlag i personvernforordninga (sjå «Behandlingsgrunnlag»).
For nokre prosjekt må det i tillegg til REK-søknad, også sendast søknad eller informasjon om prosjektet til andre instansar. Dette gjeld forskingsprosjekt som er underlagt
bioteknologilova, klinisk utprøving av legemiddel på menneske og klinisk utprøving av medisinsk utstyr. For medisinsk utstyr gjeld det berre utstyr som ikkje er CE-merkt, eller når CE-merkt utstyr skal brukast på nye område.
Klinisk legemiddelutprøving og klinisk utprøving av medisinsk utstyr skal i tillegg til REK, meldast til Statens legemiddelverk (SLV). Ved planlegging av kliniske studiar rår vi deg til å gå inn på NorCRIN sine sider. Alle tilsette i helseføretaka i Helse Midt-Norge kan nytte NorCRIN sine tenester.
Etter at prosjektet er godkjent av REK og eventuelle andre eksterne instansar, må du vurdere om det er krav til registrering av prosjektet i den internasjonale databasen Clinicaltrials.gov før første pasient blir inkludert i studien. Slik registrering kan vere en føresetnad for seinare publisering. Kontakt Klinisk Forskningsenhet Midt Norge,
Klinforsk. Alle kliniske studiar i HMR skal registrerast i Clinicaltrials.gov før prosjektet kan starte opp. HMR har konto i ClinicalTrials.gov.
Nokre innovasjonsprosjekt kan bli til forskingsprosjekt. Det er viktig å ta høgde for dette i planlegginga. Ta kontakt med personvernrådgjevar for forsking i HMR for avklaring kring innovasjonsprosjekt.
Andre typar prosjekt som nytter personopplysingar, men som ikkje er definert som medisinsk og helsefagleg forsking, treng berre intern forankring i eiga verksemd og lovleg behandlingsgrunnlag i personvernforordninga. I HMR vil du gjennom saksbehandling i DAC få nødvendig behandlingsgrunnlag.
Døme på forsking/prosjekt som normalt ikkje er definert som medisinsk og helsefagleg forsking, er samfunnsforsking, helsetenesteforsking, kvalitetsforbetringsprosjekt, register- og innovasjonsprosjekt (utanfor REK sitt mandat).
Dersom du vil hente data frå eksterne kjelder (eks. nasjonale kvalitetsregister eller sentrale helseregister), trengst det godkjenning frå kvar einskild registerforvaltar. Ver merksam på at henting av registerdata kan ta tid og at det har ein kostnad. Godkjenning frå eksterne instansar skal arkiverast i organisasjonen sitt saksbehandlingssystem.
Det kan vere nødvendig med godkjenning frå Helsedirektoratet ved forsking knytt til bioteknologi som t.d. fosterdiagnostikk og genetiske undersøkingar.
Først når alle godkjenningar som trengst er innhenta, og behandling av opplysingar i prosjektet har eit gyldig behandlingsgrunnlag, kan innsamling av data starte. Prosjektleiar har ansvar for at alle forskingsdata, personopplysingar, humant biologisk materiale - og eventuell koplingsnøkkel til persondata er forsvarleg oppbevart. Dette inneber å undersøke om den tiltenkte lagringsløysinga tilfredsstiller verksemda sine krav til sikker lagring.
Hovudregelen er at forskingsdata skal oppbevarast avidentifisert, det vil seie at direkte identifiserande element (til dømes fødselsnummer) og andre forskingsdata skal lagrast kvar for seg. Det er då vanleg å opprette ein koplingsnøkkel der ein kode knyt direkte identifiserande opplysingar til andre data.
Når både data og koplingsnøkkel vert lagra elektronisk, er det krav om lagring på skilde område, og koplingsnøkkelen må være spesielt sikra, til dømes kryptert. Det er svært sjeldan at forskargruppene har tilgang til koplingsnøkkel. Når personopplysingar og/eller koplingsnøkkel vert lagra på papir, må prosjektet forsikre seg om at koplingsnøkkel og opplysingar er lagra på skilde område og oppbevart på tilgangsstyrte område (nøkkel, nøkkelkort, skap/skuffer med lås). Opplysingar som blir handtert og lagra skal vere relevante og nødvendige for forskingsprosjektet sitt føremål slik det er godkjent av REK og andre relevante instansar.
Opplysingane som blir behandla skal vere tilstrekkelege, relevante og avgrensa til det som er nødvendig for føremålet med behandlinga og graden av personidentifikasjon skal ikkje vere større enn det som er nødvendig for det aktuelle føremålet.
Alle tilsette har ansvar for å gjere seg kjende med og etterleve krava til eiga rolle i styringssystemet for informasjonstryggleik. Tilsette har ansvar for å bidra til kontinuerleg forbetring av informasjonstryggleiksarbeidet gjennom å melde frå om avvik.
Innsamlings-/datalagringsløysingar Helse Midt-Norge har etablert
Helseforskingsportalen som gir forslag for sikker innsamling, deling, analyse og lagring av forskingsdata. Løysingane har godkjende risikovurderingar for Helse Midt-Norge. Portalen har filterfunksjon som hjelper deg til å finne løysingane som høver til ditt forskingsprosjekt. Med unntak for multisenterstudiar med eigne lagringsløysingar, er det løysingane i Helseforskingsportalen vi skal bruke for studiar som nytter data frå HMR.
Dersom ei anna verksemd skal behandle data på vegne av helseføretaket, må det underskrivast en databehandlaravtale med den eksterne verksemda. Ta kontakt med FIUK for rettleiing. I kliniske forskingsprosjekt kan du nytte NorCRIN sine prosedyrar for datahandtering og –avtalar.
Gjennomføring
Sjekkliste før gjennomføring:
• Forankring i eigen klinikk/stabsavdeling • Pasientgrunnlag og fasilitetar for gjennomføring av forskinga,
sjå eiga sjekkliste • Intern godkjenning frå DAC • Databehandlaravtale dersom anna verksemd skal behandle data frå helseføretaket
• Val av godkjend lagringsløysing • Registrert i Clinical Trials, dersom aktuelt • Godkjenning frå REK, SLV eller Helsedirektoratet når aktuelt • Gjort greie for DPIA • Signert samtykke frå deltakarane (så lenge REK ikkje har gitt fritak frå teieplikt)
• Godkjenning frå registerforvaltar ved henting av data frå eksterne kjelder
Større endringar i føremål, metode, tidsløp eller organisering skal godkjennast av REK, eventuelt av dei andre instansane som opphavleg godkjende prosjektet. Prosjektleiar må skrive ein grunngiven søknad. Det skal også sendast endringsmelding til DAC på ordinært søknadsskjema. Det er viktig å bruke saksnummeret som forskingsprosjektet blei tildelt ved første godkjenning i DAC. DAC gjer ei vurdering om føresetnadene for endring eller forlenging er til stades. Ved store endringar som vedkjem forskingsdeltakarane direkte, kan det bli krav om at det blir innhenta nytt samtykke, eventuelt at dei skal informerast skriftleg på nytt.
Deltaking i forskingsprosjekt er som hovudregel frivillig. Den som har gitt samtykke, kan krevje innsyn og korrigering av urette opplysingar. Vedkomande kan trekke seg frå vidare deltaking eller trekke tilbake samtykke. Dette må kome fram i prosjektet sitt informasjonsskriv. Deltakarar i forskingsprosjekt har også rett til innsyn og informasjon om behandlinga av opplysingar i prosjektet. Sjå nærare omtale om samtykke i ordlista (lenke).
Avvik og uønskte hendingar i denne samanhengen gjeld hendingar knytt til forskingsdeltakarar eller avvik frå godkjenningane som er gitt. Uønskte hendingar skal straks meldast til forskingsansvarleg, enten via prosjektleiar eller direkte, av den som oppdagar hendinga. Avvik i høve brot på personvernregelverket skal handterast i tråd med det lokale helseføretaket sine avviksprosedyrar.
Avviksmeldingar og tiltak som er gjort for å lukke avvika, skal dokumenterast i saksbehandlingssystemet.
For uønskte hendingar under forskinga skal
EQS kvalitetssystem brukast. På
søknadsskjema, gi opp namn på prosjekt og definer emne for hendinga. Standardrutinar for avviksmelding skal følgjast opp av linjeleiar.
Forskingsprosjektet skal stoppe til nærare avklaring er gjort med den forskingsansvarlege og REK dersom det oppstår tvil om prosjektet er forsvarleg og risikoen er uakseptabel. Korleis ein skal handle i slike situasjonar, må være omhandla i forskingsprotokollen.
Prosjektleiar eller forskingsansvarleg skal melde alvorlege, uønskte og uventa medisinske hendingar til Statens helsetilsyn. Ved unaturlege dødsfall skal politiet varslast omgåande. Prosjektleiar skal straks informere forskingsdeltakarar dersom dei er blitt påført skade, eller det har oppstått komplikasjonar som følgje av forskingsprosjektet. Samtidig skal forskingsdeltakaren bli orientert om høvet til å søke erstatning hos Norsk Pasientskadeerstatning (NPE).
Opphavsrett til eige vitskapleg arbeid. Som utgangspunkt eig forfattar sjølv rettar til eige arbeid, jf. §2 i den norske
Åndsverkloven. Dette inneber at arbeidet er verna av opphavsrett dersom arbeidet bl.a. er litterært, vitskapeleg i form av artikkel, ei avhandling i uttrykt form av skrift eller illustrasjon. Arbeidet må også vere originalt og kreativt (eit åndsverk). Opphavsretten varar normalt i 70 år etter opphavspersonens død
Medforfattarskap Grunnlaget for medforfattarskap i opphavsretten er bidrag til utforming av verket. Det betyr i praksis at alle forfattarane må ha gitt bidrag til skriving av ein tekst. I vitskapeleg publisering er dei etiske reglane for medforfattarskap ofte annleis enn dei som er stadfesta av opphavsretten. Mange fagmiljø held seg til
Vancouver-reglane (s.2) for medforfattarskap. Reglane inneber at alle forfattarane må ha gitt monalege bidrag til: • planlegging, utforming, innsamling av data eller analyse og tolking av data, • utarbeiding av sjølve manuskriptet eller kritisk revisjon av innhaldet, • godkjenning av endeleg manuskriptversjon. Alle tre vilkåra må være oppfylt.
Adressering Alle artikkelforfattarane må oppgi helseføretak og klinikk. Dette er m.a. viktig for at føretaket skal få forskingspoeng (økonomiske kompensasjon) og oppfylle krav til forskingsaktivitet. Adressering av føretaket i engelskspråklege tidsskrift: Møre and Romsdal Hospital Trust, Norway.
Avslutning
Bruk av helse- og personopplysingar i forsking er av mellombels art. Dersom REK ikkje har godkjent vidare oppbevaring, skal prosjektleiar sørge for at forskingsdata blir anonymisert eller sletta og humant biologisk materiale destruert når prosjektet er ferdig. Kopiar av data må handterast på same måten. Ein kopi av fullstendig anonymiserte data kan behaldast. Anonymisering vert vanlegvis gjort ved å slette koplingsnøkkelen. Viss dette ikkje er utført, må alle direkte og indirekte person-identifiserbare opplysingar fjernast frå forskingsdata. Derfor kan det vere ein ide å ta eventuelle ønskje om forlenga lagring av forskingsdata med i REK-søknaden.
Unntak Forskingsdata og tilknytte dokument skal ikkje slettast dersom tilsynsmyndigheit har opne saker knytt til forskingsprosjektet. Dette gjeld også om prosjektleiar eller medarbeidarar er under gransking, t.d. av tilsynsmyndigheit, «Nasjonalt utvalg for gransking av uredelighet i forskning» eller eit av Redeligheitsutvala på ulike universitet og høgskular. Ved langtidslagring i samband med klinisk legemiddelutprøving, utprøving av medisinsk teknisk utstyr (ikkje CE-merkt eller nytt bruksområde), gjeld også andre og pålagde oppgåver. Essensielle dokument, forskingsdata/Case Report Forms (CRF), kodelister og visse dokument skal lagrast i minimum 15 år etter at studien er avslutta. Lengre oppbevaring er aktuelt dersom anna regelverk ligg til grunn (t.d. somatisk celleterapi eller genterapi). Sjå Norcrin.no for nærare informasjon.